

{kind=link}
{kind=link}
{kind=link}
两株盖塔病毒全基因组序列测定与分子遗传进化
[李彬1, 2# , 付士红2# , 查冰2 , 范娜2 , 李元元2, 3 , 徐学平1, 2 , 应思超1, 2 , 聂凯2 , 许松涛2 , 李兴洲1, *  , 王环宇
, 王环宇2, * , 梁国栋2, * ]
, 王环宇, 梁国栋]
|
|


作者简介:李彬(1998—),女,在读硕士,研究方向:虫媒病毒监测与研究。 付士红(1967—),女,本科,副主任技师,研究方向:虫媒病毒监测与研究。
#付士红为共同第一作者
目的 对2011年和2017年分别在我国甘肃省和海南省分离的两株盖塔病毒(GS11-155和HNDZ1712-1)进行全基因组核苷酸序列测定,分析其与我国1964年首次分离的盖塔病毒(M1)的分子差异及分子遗传进化特征。方法 使用病毒基因扩增技术测定新分离的两株盖塔病毒全基因组核苷酸序列,建立盖塔病毒基因组数据集并使用生物信息学软件进行病毒分子特征及分子遗传进化分析等。结果 两株新分离盖塔病毒(GS11-155和HNDZ1712-1)基因组全长分别为11 690 nt和11 621 nt。两株病毒均具有甲病毒基因组结构特征。虽然两株病毒的结构基因、非结构基因以及非编码的连接区核苷酸序列长度均完全相同,但是病毒基因组5'和3'非编码区的核苷酸序列长度存在差异。两病毒株基因组3'UTR重复序列单元的结构未发生变化。病毒基因组同源性分析结果显示,海南省2017年分离的盖塔病毒(HNDZ1712-1)与我国于1964年首次在海南省蚊虫分离的盖塔病毒(M1)之间的核苷酸与氨基酸同源性分别为97.7%和98.1%。盖塔病毒全基因组核苷酸序列的分子遗传进化分析结果显示,本研究的两株病毒及其所属病毒株分别形成两个单独的进化簇,甘肃2011年进化簇和海南2017年进化簇,并与1964年海南首次分离的病毒形成完全独立的进化种群。结论 虽然HNDZ1712-1病毒同样分离自海南岛蚊虫标本,但是与1964年海南岛分离的病毒(M1)处在完全不同的进化分支,而与数千公里之外的甘肃省分离株(GS11-155)具有较近的进化关系,提示新分离的两株盖塔病毒与1964年分离的盖塔病毒存在较大分子遗传差异。
, WANG Huan-yu, LIANG Guo-dong
Objective The main aim of the study is to sequence the complete genome of two Getah virus strains (GS11-155 and HNDZ1712-1) isolated in Gansu Province and Hainan Province in 2011 and 2017 respectively and analyze the molecular and genetic evolution of the two strains compared with M1, which was first isolated in 1964 in Hainan Province, China.Methods Genome of two newly isolated Getah viruses were sequenced by virus gene amplification technique, and the genomic database of Getah viruses was established. The molecular characteristics and genetic evolution of the viruses were analyzed by bioinformatics software.Results The genome length of two new isolated Getah virus strains (GS11-155 and HNDZ1712-1) was 11 690 nt and 11 621 nt, respectively. Both strains had the structural characteristics of Alphavirus genome. Although the nucleotide sequence lengths of structural genes, non-structural genes and non-coding junction regions of the two strains were identical, the nucleotide sequence lengths of the 5' and 3' non-coding regions of the viral genomes were a few different. The 3'UTR repeats elements in the genomes of the two virus strains did not change. It was 97.7% and 98.1% different of nucleotide and amino acid homology between both strains of Getah virus, HNDZ1712-1 isolated in 2017 and M1 isolated in 1964 in Hainan Province. Interesting, Gansu 2011 cluster and Hainan 2017 cluster were emerged leading by both strains GS11-155 and HNDZ1712-1 respectively, those two clusters totally independent with M1 virus isolated from Hainan in 1964 in whole genome phylogenetic analysis first.Conclusions Although the HNDZ1712-1 was also isolated from mosquito samples in Hainan Province, it was in a completely different evolutionary branch from the M1 isolated from Hainan Island in 1964, and was closely related to the strain isolated from Gansu Province (GS11-155) thousands of kilometers away. It is suggested that the two new strains of Getah virus are different from the Getah virus isolated in 1964.
盖塔病毒(Getah virus, GETV)隶属于披膜病毒科(Togaviridae)甲病毒属(Alphavirus)[1]。盖塔病毒于1955年首次在马来西亚采集的雪背库蚊(Culex gelidus)标本分离获得, 其原株型为MM2021, 此后发现盖塔病毒为蚊虫传播的虫媒病毒[1]。盖塔病毒感染可以引起家畜动物疾病, 如马的发热、皮疹、后腿水肿及淋巴结肿大, 并能引起猪的流产等[2, 3], 因此盖塔病毒被认为是重要的动物源性病原体。日本在1970—1980年发生多次盖塔病毒感染引起的马匹和猪的疾病流行[2, 3, 4, 5, 6]。特别是2014年日本报道盖塔病毒感染引起的2 000匹赛马马匹的疾病流行, 采集到的49份发热马的血清标本中有25份盖塔病毒基因检测阳性, 基因检测阳性率达51.02%(25/49)[7]。1990年印度发生大量马匹盖塔病毒感染的暴发流行[8]。我国近10年也已经发现在饲养的猪和蓝狐中发生盖塔病毒感染的暴发流行[9, 10]。可见盖塔病毒感染对畜牧业生产产生巨大疾病负担。
盖塔病毒呈圆形, 具有包膜, 为单股正链RNA [singlestranded(ss)RNA]病毒。病毒基因组5'端具有甲基化(7-methylguanosine)帽子结构, 而3'末端有数量不等的poly(A)尾。病毒基因组具有两个开放编码区(open reading frames, ORFs), 分别编码非结构基因和结构基因, 其中病毒基因组5'端前2/3部分编码病毒非结构蛋白(non-structural protein), 非结构蛋白(包括nspl到nsp4)负责病毒RNA转录和复制、多聚蛋白质的切割和RNA带帽过程(capping)。病毒基因组后1/3部分为病毒结构基因, 其26S RNA(subgenomic RNA)mRNA编码病毒多个结构蛋白, 包括C(Capsid, 衣壳蛋白)、E3、E2、6K和E1。E蛋白(包膜蛋白)是病毒与宿主细胞接触、并引起疾病和免疫反应的主要蛋白质[11]。
我国自1964年首次在海南省蚊虫分离到盖塔病毒(M1)[12], 以后陆续在中国多个省市的蚊虫甚至动物标本中鉴定出多株盖塔病毒[13, 14, 15, 16, 17, 18, 19, 20, 21, 22], 但是缺少对新分离盖塔病毒与我国1964年首次分离的盖塔病毒(M1)之间进行全基因组水平的比较, 包括病毒全基因组分子生物学特征的比较和全基因组水平的分子遗传进化分析等。为此, 本研究首先对2011年在甘肃省三带喙库蚊中分离的盖塔病毒GS11-155[15]以及2017年在海南省三带喙库蚊中分离的盖塔病HNDZ1712-1[16]进行全基因组序列测定, 在此基础上将两株病毒与我国1964年首次分离的盖塔病毒(M1)进行全基因组水平的比较分析, 以期了解我国目前流行的盖塔病毒与50余年前的盖塔病毒之间的分子遗传差异等, 为盖塔病毒在我国动物中的感染与预防控制提供科学依据。
本室保存的两株盖塔病毒GS11-155和HNDZ1712-1病毒分离株分别分离自我国甘肃省和海南省采集的三带喙库蚊标本[15, 16]。以上两株病毒分别在BHK细胞复苏培养, 收集细胞感染液用于病毒基因组核苷酸序列测定。
使用RNA提取试剂盒Viral RNA mini Kit(QIAamp; Qiagen, Valencia, CA)分别提取GS11-155和HNDZ1712-1病毒感染的BHK-21细胞上清中的总RNA。使用Ready-To-Go kit试剂盒中的第一链反应管(GE Healthcare, Little Chalfont, Buckinghamshire, UK)与随机引物pd(N)6(TaKaRa, Japan)制备病毒RNA的cDNA文库[14]。
利用文献[23]盖塔病毒13对扩增引物对盖塔病毒基因组扩增, 阳性PCR产物送天一辉远生物技术有限公司(中国北京), 使用四色荧光标记双脱氧终止法对阳性PCR产物进行测序[24, 25]。
从GenBank数据库中收集不同国家、不同吸血昆虫和不同宿主动物标本中分离的盖塔病毒基因序列, 使用BioEdit(version7.0, Thomas)进行基因多序列比对; 使用MEGA-X软件完成基于Neighbour-Joining (NJ)方法的系统进化分析, 选择Tamura-Nei为最适替换模型。Bootstrap值设定为1000, 使用MegAlign软件(DNAStar, Madison, WI)进行核苷酸和氨基酸序列同源性分析[16]。
GS11-155和HNDZ1712-1分别接种BHK细胞, 连续培养3 d后出现明显细胞病变(CPE), 与对照细胞相比病变细胞表现为细胞生长缓慢, 细胞圆缩和脱落。提取病毒感染上清中的RNA, 制备病毒基因组cDNA, 进行扩增和测序。
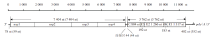
甘肃省分离的GS11-155病毒和海南省分离的HNDZ1712-1病毒全基因组核苷酸序列分别为11 690 nt和11 621 nt。两株病毒非结构基因均为7 404 nt, 各编码4种非结构蛋白(nsp1~nsp4)。两株病毒结构基因全长均为3 762 nt, 各自编码5个结构蛋白(C、E3、E2、6K、E1)。两病毒株结构基因与非结构基因之间均各有44 nt的连接区序列。两株病毒5'末端非编码区(5'UTR)核苷酸序列分别为78 nt和59 nt, 而病毒3'UTR核苷酸序列长度分别为402 nt和352 nt。两株盖塔病毒基因组核苷酸序列长度及各功能基因结构见图1。
利用来自GenBank的病毒基因序列信息, 构建盖塔病毒全基因组核苷酸序列(11 210 nt)以及E2基因核苷酸序列(1 266 nt)系统进化数据集(表1), 使用邻接方法分析甘肃省和海南省新分离的两株盖塔病毒的分子遗传进化状况(图2)。无论病毒全基因组或E2基因核苷酸序列进化分析结果均显示, 55株盖塔病毒分为4个基因型(G1~G4)。基因1型仅含1株病毒, 即1955年在马来西亚蚊虫分离的盖塔病毒(MM2021); 基因2型含两株为1956年在日本分离的盖塔病毒; 基因4型含4株病毒, 分别为2012年中国云南省分离株(YN12031)、2000年俄罗斯分离株(LEIV/16275/Russia)、2012年马来西亚分离株(B254)和2017年泰国分离株(SW)。可见基因1、2、4型盖塔病毒仅占全部毒株的13%(7/55), 其余毒株均属于基因3型。基因3型病毒进化分支中, 1964年海南省分离的M1分离株为独立的进化分支, 而与之对应的进化分支(47株病毒)包括中国、日本、韩国及蒙古国于1964年以后分离的盖塔病毒分离株, 可见基因3型盖塔病毒为优势基因型病毒种群。见图2。
| 表1 本研究使用盖塔病毒毒株基因序列信息 Table 1 Genetic sequence information of Getah virus strain was used in this study |
进一步分析发现, 虽然2011年甘肃省和2017年海南省分离的盖塔病毒(GS11-155和HND1712-1)均属于基因3型病毒, 但是这两株病毒及其所属病毒分离株分别形成两个单独的进化簇, 即甘肃2011进化簇和海南2017进化簇。而两个进化簇在病毒全基因组和E2基因的分子遗传进化树中的位置截然不同。全基因组分子遗传进化分析显示(图2A), 甘肃2011年进化簇中包含20株病毒, 而且海南2017年进化簇的病毒被包括在甘肃2011进化簇中。而E2基因分子遗传进化分析显示(图2B), 2011甘肃和2017海南及其所属病毒株分别成为两个不同的进化簇, 甘肃2011年进化簇中含有12株病毒, 而海南2017年进化簇含有8株病毒。
甘肃省2011年分离株(GS11-155)与海南省2017年分离株(HND1712-1)核苷酸同源性98.8%, 氨基酸同源性99.3%。而以上两株新分离盖塔病毒与我国1964年在海南省最先分离的盖塔病毒(M1)分析结果显示, 3株病毒E2基因核苷酸和氨基酸同源性在97.7%~98.8%和98.1%~99.3%。进一步分析发现, 海南省2017年分离株(HND1712-1)与1964年分离株(M1)之间核苷酸和氨基酸同源性分别为97.7%和98.1%。同样海南省1964年分离株(M1)与数千公里外的甘肃省2011年分离株(GS11-155)核苷酸同源性98.1%, 氨基酸同源性98.8%。
甘肃省和海南省分离的盖塔病毒(GS11-155与HNDZ1712-1)的3'UTR的分子特征分析结果发现, 两株病毒均包含盖塔病毒特有的3个长度为60 nt左右的重复序列单元, 3个 重复序列单元分别位于编码取终止子后1(68~122 nt); 2(126~178 nt)和3(182~247 nt)。GS11-155与HNDZ1712-1病毒的3个重复序列单元与盖塔病毒马来西亚株MM2021完全相同(图3)。结果提示本研究分离测定的两株盖塔病毒属于甲病毒属盖塔病毒。
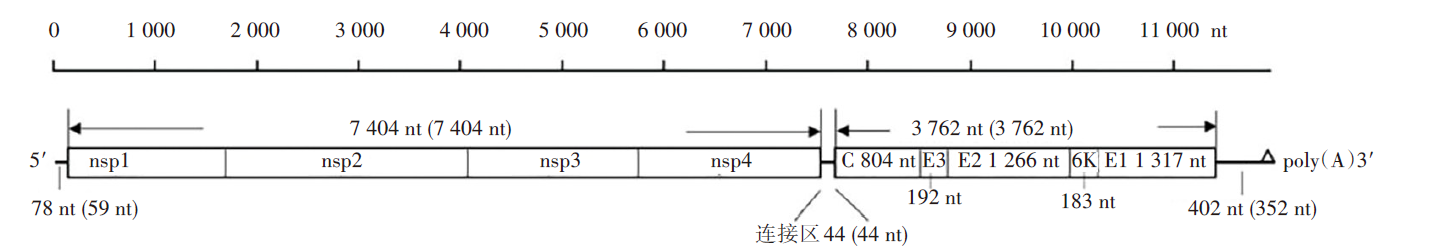 | 图1 盖塔病毒(GS11-155和HNDZ1712-1)全基因组结构 图中括号内外分别为GS11-155病毒和HNDZ1712-1病毒基因组核苷酸序列长度。Fig. 1 Complete genome structure of Getah virus (GS11-155 and HNDZ1712-1) Inside and outside parentheses are the nucleotide sequence lengths of GS11-155 and HNDZ1712-1 genomes respectively. |
 | 图2 盖塔病毒系统进化分析 A为全基因组进化树; B为E2基因进化树; ●表示本研究盖塔病毒; ▲为1964年海南分离株M1。 |
Referral and tracing of tuberculosis cases online reported by nontuberculosis bodies
A is the whole genome evolutionary tree; B is the E2 gene evolutionary tree; ● represents Getah virus in this study; ▲ represents 1964 Hainan strain M1.
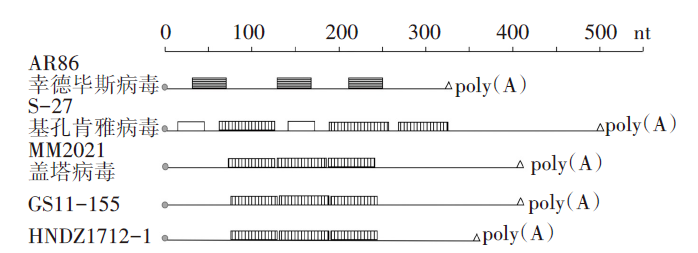 | 图3 盖塔病毒3′ UTR重复序列结构图 圆圈表示结构蛋白基因区域的终止密码子; 不同类型的方框表示不同的重复序列; 三角代表病毒基因组3′末端的Poly(A)尾。Fig. 3 Structure of Getah virus 3′UTR repeat sequence The circles in the figure indicate the stop codon of the structural protein gene region. Different types of boxes represent different sequences of repeats; the triangle represents the Poly(A) tail at the 3′ end of the viral genome. |
本研究分别对甘肃省2011年和海南省2017年采集的三带喙库蚊标本中分离的盖塔病毒GS11-155和HND1712-1进行了全基因组核苷酸序列测定与分析, 以期比较以上两株病毒之间及其与1964年在我国首次分离的盖塔病毒(M1)在病毒全基因组水平的差异。三株病毒虽然分离时间间隔50余年, 分离地域相隔数千公里, 但是无论病毒基因组长度, E2基因核苷酸和氨基酸同源性, 或者病毒3'UTR重复序列单元的结构等均未发生较大变化。以上病毒均属于基因3型盖塔病毒, 但是1964年分离的病毒株处于基因3型盖塔病毒分子进化的根, 属于基因3型病毒中最古老的病毒, 而甘肃和海南省新分离毒株与其所属病毒形成单独的进化簇, 与M1病毒完全独立, 可见海南省2017年与甘肃省2011年新分离的盖塔病毒与1964年分离的病毒在病毒基因组分子遗传进化存在较大差异。
利用病毒基因组核苷酸序列和生物信息学方法对盖塔病毒基因型变迁的研究结果显示[14], 盖塔病毒大约起源于145年前(95% HPD:75~244), 属于新出现的病毒(emerging virus), 并逐渐演化出4个基因型。基因1型病毒为1955年马来西亚分离的盖塔病毒(盖塔病毒的原型病毒MM2021病毒株), 位于盖塔病毒基因系统发育树的根部, 提示该病毒为最古老的盖塔病毒分离株。基因2型病毒出现在大约60年前(95% HPD:59~73), 包括1956年在日本分离的两株病毒(sagiyama virus)。基因3型病毒大约出现在50年前(95% HPD:51~72)。基因3型盖塔病毒可以分为两个进化分支, 其一为1964年我国分离的盖塔病毒(M1)也是基因3型病毒进化分支中最古老的病毒, 所有其它基因3型病毒与M1病毒完全独立并平行进化出基因3型病毒第二进化分支。基因3型病毒第二进化分支的病毒株最多, 包括1960年代年至2020年在中国多省份分离的病毒株以及日本、韩国和蒙古国分离的盖塔病毒。进一步分析发现, 这一进化分支中不仅包括日本和蒙古国及中国多个省份分离的病毒株, 而且这些毒株又可以分为毒株数量不等的进化簇, 包括甘肃2011年进化簇和海南2017年进化簇等。基因4型盖塔病毒为最年轻的盖塔病毒种群, 大约在30年前出现(95% HPD:16~55), 基因4型病毒含4株病毒, 分别为2012年中国云南省分离株(YN12031)、2000年俄罗斯分离株(LEIV/16275/Mag)、2012年马来西亚分离株(B254)和2017年泰国分离株(SW)。由此可见基因3型盖塔病毒为优势基因型病毒, 引领了盖塔病毒的地域扩张和分子遗传进化趋势。
鉴于盖塔病毒已经成为在亚洲地区普遍存在的蚊传虫媒病毒, 因此盖塔病毒的时空动力学分布特征, 即病毒迁徙传播路线成为被关注的科学问题。研究结果显示, 马来西亚、日本和中国云南省是盖塔病毒3个主要的播散地。盖塔病毒可能存在如下的播散路线:(1)1917年从马来西亚播散到日本; (2)1960年代从日本播散到中国海南省; (3)1970年代从日本播散到中国云南省; (4)此后以云南省为播散源头传播至我国河北、上海、甘肃和四川等内地省份[25]。结合盖塔病毒分子遗传进化结果由此可见1、2、4基因型盖塔病毒成为稳定病毒种群在局部地区流行, 而基因3型盖塔病毒的地域分布范围, 特别是在近20年来迅速扩张, 在亚洲大陆地区不断向北扩散, 传播至我国北方地区的河北省、山东省、内蒙古、甘肃省等, 并已经传播至蒙古国[25]。而基因3型盖塔病毒在传播和迁徙过程中不断分化, 演变出不同的进化簇(claster)。基因3型盖塔病毒在迁徙传播过程中不断分化成为优势基因型病毒种群。
盖塔病毒和乙脑病毒在我国的迁徙和传播路线具有相似之处。乙脑病毒起源地为马来西亚/菲律宾等东南亚地区[26], 此后在我国云南省形成乙脑病毒在亚洲大陆的起源地, 并形成3条迁徙路线, 东部迁徙路线从云南省向东传播至我国东南沿海地区(福建、海南、浙江)和日本与韩国; 中部传播路线向湖南、湖北、华北地区传播; 西部迁徙路线从云南省传播至我国四川、甘肃等地[27]。以上结果可能具有一定的巧合性, 但是2011年甘肃和2017年海南分离的盖塔病毒的分离地均处在乙脑病毒和盖塔病毒迁徙传播路线中。虽然乙脑病毒和盖塔病毒是否存在相同的迁徙途径还需要开展更细致的分析与研究, 但是乙脑和盖塔病毒在一些地区的并存是不争的事实, 无论海南省或者甘肃省均多次分离到乙脑病毒和盖塔病毒[12, 15, 16], 我国其他地区也存在同样情况[28]。
自1970年代, 中国在首次分离到盖塔病毒的海南岛地区采集的不明原因发热患者血清标本的盖塔病毒抗体阳性率为26.4%(24/91), 而当地健康人血清中抗体阳性率为3.4%(2/58), 可见当地不明原因发热患者中可能存在盖塔病毒感染[12]。但是尚未发现盖塔病毒感染引起的临床病例。此外, 澳大利亚[29]、马来西亚[30]也在当地健康人血清标本中检测到盖塔病毒抗体阳性。如前所述, 日本、印度等国已经发生多次盖塔病毒在动物中的流行, 造成巨大经济损失[1, 4, 5, 6, 7, 8]。值得注意的是, 我国虽然1964年即从蚊虫分离到盖塔病毒[12], 但是盖塔病毒在动物中引起疾病流行是近10年的事情。2017年6—7月, 中国湖南省的一个养猪场出现盖塔病毒感染的爆发流行。大约200头小猪在出生后5~10 d死亡, 150多头怀孕母猪在这次疫情中死亡[9]。2017年9月我国东部的山东省某农场饲养的25只5个月大的蓝狐出现突然发热、厌食和抑郁的症状。25只动物中6只出现神经症状, 并在发病第3天死亡[10]。2018年8月, 在中国南方广东省的一个马术训练中心, 观察到赛马的突然发烧与盖塔病毒感染有关[19]。此外, 自2011年以来我国已经在河南[17]、湖南[9]、山东[10]、广东[19]、吉林[20]、广西[21]和四川[22]等省份发生盖塔病毒在动物感染的流行。本研究结果显示, 甘肃2011年进化簇中含有日本2014年和2015年引起盖塔病毒流行的分离株14-I-605, 15-I-752。海南2017年进化簇中含有2018和2020年四川小熊猫感染的病毒分离株(GETV/SCrph328/2018和GETV/SCrph129/2020)。因此, 加强我国盖塔病毒分子遗传特征研究, 特别是及时发现盖塔病毒对动物感染疾病的流行具有十分重要的意义。
利益冲突声明 所有作者声明不存在利益冲突
编辑:王佳燕
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|