{kind=link}
156例疑似罕见遗传代谢病患儿的个体化精准诊断分析
[田露, 逯军*  ]
]
]
|
|


作者简介:田露(1998—),女,硕士研究生,研究方向:儿童罕见遗传代谢病。
目的 探讨疑似罕见遗传代谢病(inherited metabolic disorders, IMD)患儿的精准诊断,并对比质谱技术与基因检测技术在罕见IMD诊断中的应用价值。方法 回顾性分析2017年3月至2021年12月就诊于中南大学湘雅医学院附属海口医院儿科疑似罕见IMD患儿的临床信息、质谱与基因结果。结果 156例疑似罕见IMD患儿均行质谱检测,初检阳性67例,阴性89例。初检阳性患儿均进行复检,复检阳性19例。复检阳性中13例行基因检测,检出基因阳性9例、阴性4例。初检阴性中治疗疗效欠佳、临床仍高度怀疑IMD的54例患儿完善基因检测,检出阳性15例、阴性39例。将两种检测方法的结果进行对比,质谱检测阳性率为19.4%(13/67),基因检测阳性率为35.8%(24/67)。连续性修正 χ2检验提示基因与质谱检测结果有差别,差异有统计学意义( P<0.05)。以基因检测为金标准,质谱检测敏感度为37.5%(95% CI:19.6%~59.2%),特异度为90.7%(95% CI:76.9%~97.0%)。24例确诊患儿中,5例经基因panel确诊,19例经全外显子测序(whole gexome sequencing,WES)确诊。其中1例经WES确诊的患儿,确诊前完善基因panel未检出致病变异。检测发现 DNM1L基因c.1040C>G及 AMN基因c.651+1G>C为疾病致病基因新发变异位点,具有临床意义。结论 质谱技术诊断IMD能力有限,对疑似罕见IMD患儿可首选基因检测尤其是WES进行个体化诊断。此外本研究中WES发现的新发变异位点,丰富了致病基因突变谱,为进一步功能生物学实验提供方向。
Objective To explore the accurate diagnosis of children with suspected rare inherited metabolic diseases, and to compare the application value of mass spectrometry and genetic testing in the diagnosis of rare inherited metabolic diseases (IMD).Methods The clinical information, mass spectrometry, and genetic results of children with suspected rare inherited metabolic diseases admitted to the Department of Pediatrics, the Affiliated Haikou Hospital of Xiangya Medical College, Central South University from March 2017 to December 2021 were analyzed retrospectively.Results 156 children with suspected rare inherited metabolic diseases were detected by mass spectrometry, 67 cases were positive and 89 cases were negative. Children with positive initial examination were retested, and 19 cases were positive. Among the retest positive cases, 13 cases were given genetic testing, and 9 cases were positive and 4 cases were negative. Among the initial negative cases, 54 children with poor therapeutic effect and high clinical suspicion of inherited metabolic diseases completed genetic testing, 15 cases were positive and 39 cases were negative. The results of the two detection methods were compared, the positive rate of mass spectrometry was 19.4%(13/67), and the positive rate of genetic testing was 35.8%(24/67). The continuity correction of Pearson's chi-square test of continuity correction suggested that the results of genetic testing and mass spectrometry were different, and the difference was statistically significant ( P<0.05). Taking genetic testing as the gold standard, the sensitivity and specificity of mass spectrometry detection were 37.5% (95% CI:19.6%-59.2%) and 90.7% (95% CI:76.9%-97.0%), respectively. Among the 24 confirmed cases, 5 cases were diagnosed by gene panel and 19 cases were diagnosed by whole exome sequencing (WES). One case diagnosed by WES had no pathogenic mutation detected by gene panel before diagnosis. The detection of DNM1L gene c.1040C>G and AMN gene c.651+1G>C are novel pathogenic gene variants, which have clinical significance.Conclusions The ability of mass spectrometry in the diagnosis of inherited metabolic diseases is limited. Genetic testing, especially whole exome sequencing, can be the first choice for individualized diagnosis of suspected rare inherited metabolic diseases. In addition, the new mutation sites found by WES in this study enriched the pathogenic gene mutation spectrum and provided direction for further functional biological experiments.
遗传代谢病(inherited metabolic disorders, IMD)是指基因致病性变异导致代谢途径缺陷, 使代谢底物或中间产物异常累积和/或产生必要物能力降低的一大类单基因遗传病。IMD多为罕见病, 但总体发病率高, 约为1/13 250~1/1 178[1, 2, 3, 4, 5]。该类疾病的诊断周期从数月到数十年不等, 平均确诊时间约为4~5年, 误诊误治给患儿及家庭带来沉重负担[6, 7]。目前绝大多数IMD无根治方法, 但部分疾病如尿素循环障碍、脂肪酸代谢病、碳水化合物代谢病、有机酸血症、线粒体病、免疫缺陷病等可早期通过特定饮食和药物进行干预治疗, 有效防止病情进展, 降低发病频率及致死率, 因此迅速诊断至关重要。串联质谱检测技术(tandem mass spectrometry, MS/MS)、气相色谱质谱技术(gas chromatography-mass spectrometry, GC/MS)和基因检测技术凭借一次实验可检测多种疾病的优势广泛应用于IMD诊断。随着分子生物学技术蓬勃发展, 基因检测技术, 特别是全外显子测序(whole gexome sequencing, WES), 为罕见IMD的个体化医疗和精准诊断提供了强有力的支持[8]。同时精准诊断还要求临床医生在诊疗过程中识别出疑似IMD患儿, 予以具有诊断意义的基因检测方法, 最后依据患儿的临床资料结合基因报告作出最终判断[9]。本文通过对156例疑似罕见IMD患儿的诊断进行分析, 总结罕见IMD的精准诊断经验。
对2017年3月至2021年12月就诊于中南大学湘雅医学院附属海口医院儿科的156名疑似罕见遗传代谢病患儿的诊断及诊断方法进行回顾性研究。本研究已通过中南大学湘雅医学院附属海口医院伦理委员会审核和批准, 伦理批文编号:2022-(伦审)-196。本研究均获得患儿父母知情同意, 并签署知情同意书。
1.1.1 纳入标准 至少存在下列一种临床表型:不明原因的反复感染、呕吐、抽搐、意识障碍、精神行为异常、心律失常、生长发育落后、肝脾肿大、心肌病、白内障、皮肤毛发改变等, 或实验室检查发现难以解释的高氨血症、代谢性酸中毒、低血糖、贫血、肝功能损害、心肌损害、高乳酸血症、高脂血症等。
1.1.2 排除标准 符合以下任意一项:①病例资料不全无法补充者; ②存在多个系统畸形的患儿; ③存在已知染色体核型分析检查异常或其他非罕见IMD的遗传病患儿。
1.2.1 基因检测方案及选择依据 本研究涉及的基因检测方案包括:遗传代谢病基因panel、线粒体基因panel、脑白质基因panel及WES。遗传代谢病基因panel包含ABCD4、ACSF3、CD320、LMBRD1、MMAA、MMAB、MMACHC、MMADHC、MMUT、SUCLA2、SUCLG1、HCFC1、GCDH等836种遗传代谢病基因, 线粒体基因panel含37个线粒体基因, 脑白质基因panel含ABCD1、PHYH、GALT、PC、多种PEX基因等165种与遗传性脑白质病相关的基因。根据患儿临床表型、一般检查结果、质谱检测结果, 选择不同基因检测方案。
1.2.2 质谱及基因检测结果判读 质谱检测结果分2类:代谢成份异常增高或降低为阳性; 无代谢成分异常为阴性。基因检测结果分2类:检出与临床表型相符的变异, 包括致病、可能致病和临床意义不明的变异, 判为阳性; 反之为阴性。变异致病性分类参考美国医学遗传学和基因组学学会(Americal College of Medical Genetics and Genomics, ACMG)的遗传变异分类标准与指南。新发变异使用Mutation taster、PolyPhen评估序列进化保守性。疾病诊断分类按国际遗传代谢疾病分类(The International Classification of Inherited Metabolic Disorders, ICIMD) [10]。
1.2.3 资料收集与处理 收集疑似罕见IMD患儿临床信息, 包括病史、体征、辅助检查及诊治情况, 并随访。通过对患儿的诊断方法进行分析, 对比质谱技术与基因检测的诊断效能, 探讨两者在罕见IMD诊断中的应用价值。
采用SPSS 26.0统计软件、Microsoft Office Excel 2022软件对数据进行统计分析。计数资料数量用[n(%)]表示。采用Kappa一致性检验和连续性修正的χ 2检验对比质谱检测与基因检测结果的差异, P< 0.05为差异有统计学意义。
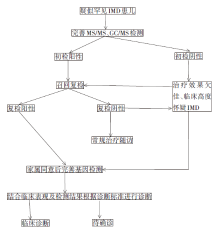
纳入的156例患儿中, 男性84例(53.8%), 女性72例(46.2%)。临床表现涉及多个系统, 神经系统包括抽搐95例(60.9%)、精神行为异常10例(6.4%)、发育迟缓21例(13.5%)、脑积水5例(3.2%)、智力低下4例(2.6%)、小头畸形3例(1.9%)及发育倒退2例(1.9%)。消化系统包括肝功能异常9例(5.8%), 呕吐5例(3.2%), 肝脾肿大2例(1.3%), 黄疸1例(0.6%)。代谢紊乱包括代谢性酸中毒7例(4.5%)、低血糖4例(2.6%)、高脂血症3例(1.9%)及高乳酸血症2例(1.3%)。肌肉系统包括肌力异常6例(3.8%)、肌酶增高15例(9.6%)。循环系统包括先天性心脏病3例(1.9%)、心律失常5例(3.2%)。血液系统包括贫血11例(7.1%)。其他有反复呼吸道感染3例(1.9%)、皮肤及毛发改变3例(1.9%)、听力及视力下降2例(1.3%)。诊断流程如图1。
 | 图1 疑似IMD患儿诊断流程图Fig. 1 Flow chart for diagnosis of children with suspected IMD |
156例患儿均进行质谱检测, 初检阳性67例, 阴性89例。初检阳性患儿均进行复检, 复检阳性19例、阴性48例。复检阳性结果中:提示某种IMD 5例, 分别为戊二酸血症I型1例, 多种酰基辅酶A脱氢酶缺乏症1例, 甲基丙二酸血症1例, 希特林蛋白缺乏症1例, 异戊酸血症1例; 提示某类或某几种IMD 6例, 分别为过氧化物酶体病2例, 脂肪酸氧化代谢疾病3例, 甲硫氨酸循环障碍性疾病1例, 尿素循环障碍性疾病1例, 短链酰基辅酶A脱氢酶缺乏症、乙基丙二酸脑病或异丁酰辅酶A脱氢酶缺乏症1例; 提示继发性代谢异常6例。
67例患儿基因检测检出阳性24例, 阴性43例。DNM1L基因c.1040C> G及AMN基因c.651+1G> C为2个检出新发变异位点经致病性分析, 前者临床意义不明, 后者为致病变异。检出29个变异位点中致病性(pathogenic, P)变异11个, 可能致病(likely pathogenic, LP)变异7个, 临床意义不明(uncertain significance, US)变异11个。阳性24例均诊断为IMD, 包含20个病种, 营养物质代谢障碍6例, 能量代谢障碍5例, 线粒体基因表达障碍1例, 脂质代谢或运输障碍6例, 杂环化合物代谢障碍1例, 复杂分子和细胞器代谢障碍2例, 辅因子及矿物质代谢障碍2例, 代谢细胞信号传导障碍1例。确诊患儿信息见表1。
| 表1 24例确诊IMD患儿的临床信息及遗传学结果 Table 1 The clinical information and genetic results of 24 children with IMD |
质谱复检阳性中提示IMD 的13例患儿进一步完善基因检测, 检出阳性9例、阴性4例。初检阴性中治疗疗效欠佳、临床仍高度怀疑IMD的54例患儿完善基因检测, 检出阳性15例、阴性39例。质谱与基因两种检测方法结果见表2。质谱检测阳性率19.4%, 阴性率80.6%; 基因检测阳性率35.8%, 阴性率64.2%。经Kappa一致性检验, Kappa值为0.314, 表明两种检测方法的一致性差。连续性修正的χ 2检验提示基因与质谱检测结果差异有统计学意义(P< 0.05)。以基因检测为标准, 质谱检测敏感度为37.5%(95%CI:19.6%~59.2%), 特异度为90.7%(95%CI:76.9%~97.0%), 阳性预测值为69.2%(95%CI:38.9%~89.6%), 阴性预测值为72.2%(95%CI:58.1%~83.1%)。
| 表2 本研究67例患儿质谱与基因两种检测方法的结果 Table 2 The results of mass spectrometry and genetic testing in 67 children |
24例确诊患儿中, 5例(20.8%)经基因panel确诊(表1), 19例(79.2%)通过WES确诊。其中1例经WES确诊的患儿, 确诊前经基因panel未检出致病变异。
IMD临床表型繁多复杂, 可累及全身所有器官系统。不同IMD的临床表型可重叠, 因此仅根据临床表型确诊较难。但出现以下情况时应警惕IMD可能[11]:①有反复流产、新生儿死亡、父母近亲婚配等家族史; ②病情呈间歇性或波动性恶化; ③不明原因反复呕吐、腹泻、厌食或喂养困难; ④难以解释的代谢紊乱和影像学异常; ⑤存在面容或肢体异常; ⑥不明原因肝脾大或心肌病; ⑦存在特殊气味或体味; ⑧存在全身受累的证据, 如发育迟缓、肌张力减退、惊厥、白内障、视神经萎缩或肾小管受累等。
临床诊断IMD的特异性实验室检查包括质谱技术、酶学活性检测和基因检测。酶学活性检测多为侵入性检查, 对标本采集和保存要求高, 检测疾病受限, 且对诊断不明的患儿难以进行针对性检测[12], 临床已少用。质谱技术仅限于辅助有机酸、氨基酸、脂肪酸氧化代谢障碍疾病的诊断。近年来, 随着基因测序成本的快速下降及生物信息学分析能力的提升, 二代测序(next generation sequencing, NGS)广泛应用于罕见病的诊断。NGS根据测序目标范围大小, 可分为基因panel测序、WES及全基因组测序(whole genome sequencing, WGS), WGS因测序成本较高, 临床少用[13]。
13例行基因检测的复检阳性患儿中, 4例为阴性。结合患儿临床资料, 未达到IMD诊断标准。由此可见质谱技术存在假阳性。假阳性出现的原因[12, 14, 15]有:第一, 代谢物浓度受饮食、营养和疾病状态的影响, 出现一过性或继发性代谢紊乱, 如肝功能异常时可引起4-羟基苯乳酸增高; 机体处于高消耗状态或营养不良时可出现氨基酸或肉碱浓度降低。第二, 代谢物浓度受药物影响, 如抗癫痫药丙戊酸钠的使用可引起尿中丙戊酸及多种有机酸增高、干扰血液中酰基肉碱的检测[16]。第三, 代谢物存在质谱技术难以辨别的同分异构体, 如检测指标C5酰基肉碱包含异戊酰肉碱、特戊酰肉碱及2-甲基丁酰肉碱等异构体[17]。
13例行基因检测的复检阳性患儿中, 9例为阳性。3例质谱与基因结果一致, 6例通过基因检测进一步明确病因和分型。其中1例患儿多次GC/MS均提示甲基丙二酸、3-羟基丙酸显著增高, MS/MS中甲硫氨酸降低, 血中同型半胱氨酸明显增高, 故诊断为甲基丙二酸尿症合并同型半胱氨酸血症。但患儿遗传代谢基因panel检测未发现基因变异, 为明确病因进一步完善WES。结果显示AMN基因存在复合杂合变异, 故修正诊断为Imerslund-Grä sbeck综合征。IGS因体内维生素B12缺乏, 影响甲硫氨酸合成酶和L-甲基丙二酰辅酶A变位酶活性, 引起甲硫氨酸降低、同型半胱氨酸及甲基丙二酸异常升高, 仅凭质谱检测易误诊[18, 19]。WES不仅从分子水平检测病因且能同时明确疾病分型, 更全面、准确作出诊断[20]。
54例行基因检测的质谱阴性患儿中, 15例通过基因结果诊断为IMD, 由此可见质谱存在一定假阴性。造成质谱假阴性的主要原因为大部分IMD不具有代谢标志物。ICIMD也指出异常代谢标志物的检出不再是诊断IMD的先决条件[10]。质谱阴性也不能排除IMD可能[21], WES是缺乏代谢标志物这类罕见IMD最有用的诊断方式[22]。
目前临床上常用的NGS技术包括基因panel及WES。基因panel针对特定疾病的致病基因进行测序, 但对于基因数庞大的非特异性表型, 如癫痫患儿在使用癫痫基因panel时, 漏诊几率高[23, 24]。WES同时对约20 000个基因外显子及其侧翼序列进行测序, 逐渐成为罕见IMD诊断的首选检测方法。基因panel的检测成本较WES小, 但WES可通过避免重复测序来降低成本[25], 近年来, NGS技术的进步使每个碱基对测序成本降至原来1/10, 极大的降低了检测费用, 为其在临床普及提供了技术支持。此外WES还可同时识别ACMG发布的对患儿护理具有医学价值的次要发现[26]。研究发现, WES不仅能提高诊断罕见IMD的能力, 还能为后续治疗与管理、遗传咨询及预防性医疗提供个性化的方案[27]。但WES也存在一定局限性, 如无法检测非编码区域的致病变异、染色体结构改变、高度同源序列区域、三核苷酸重复动态突变, 对拷贝数变异的分析能力有限等[28, 29]。有研究[30]表明, 对于父母无临床表型的家庭, 父母-先证者(Trios家系模式)测序分析可减少候选变异数, 提高诊断率, 加快精准诊断速度, 提高了达到精准诊断可能性。
本研究检出2个新发变异位点, DNM1L基因c.1040C> G变异位点为罕见变异, PubMed、gnomAD、ExAC、千人基因组及dbSNP数据库均未见收录(PM2), 多个生信软件预测均为有害变异(PP3), 经Sanger测序验证其亲生父母未发现该变异, 且患儿家族史及临床表型与新发变异特征相吻合(PS2), 故该变异为可能致病变异。AMN基因c.651+1G> C位点为经典+1的剪接突变(PVS1), PubMed、gnomAD、ExAC、千人基因组及dbSNP数据库均未见收录(PM2), 经Sanger测序验证其父母未发现该变异(PS2), 故该变异为致病变异。本文5例患儿变异位点临床意义不明, 但这些变异位点在人群中发生频率极低, 经生信分析均为有害变异, 符合家系共分离, 结合患儿症状、体征及辅助检查结果, 均考虑为IMD。
综上所述, 质谱技术诊断IMD能力有限, 对疑似罕见IMD患儿可首选基因测序尤其是WES进行个体化诊断。本研究中WES发现新发变异位点, 丰富致病基因突变谱, 为进一步功能生物学实验提供方向。
利益冲突声明 所有作者声明不存在利益冲突
编辑:朱学义 王佳燕
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|