

{kind=link}
{kind=link}
1例疑似X连锁隐性遗传病女性患儿家系遗传学分析
[王丹虹, 逯军*  ]
]
]
|
|


作者简介:王丹虹(1982—),女,硕士,副主任医师,研究方向:新生儿科方向。
目的 对1例存在染色体Xq28杂合缺失,疑似X连锁隐性遗传病的女性患儿家系进行遗传学分析以判断其发病及预后。方法 女性患儿,20 d,因“皮肤黄疸20 d,针刺部位难止血”入院,母孕晚期羊水穿刺低深度全基因组测定提示甲型血友病和X连锁智力障碍72型相关拷贝数缺失。为进一步确诊及明确预后,采集患儿及其父母外周血行基因检测,并进行染色体失活检测及遗传学分析。结果 患儿染色体Xq28 存在约 439.4 kb 的拷贝数杂合缺失变异,是ClinGen数据库收录的明确功能缺失致病编码基因。染色体失活检测显示患儿父源X染色体为极度失活偏移,单倍型分析提示受检者正常染色体遗传自母亲,父源X染色体上存在杂合缺失,故推断患儿不发病或症状轻微。结论 携带X连锁隐性遗传疾病致病性变异的女性患者有必要进行X染色体失活检测数据分析,判断其发病的可能性。
Objective Genetic analysis was performed on a female child with chromosome Xq28 heterozygous deletion and suspected X-linked recessive disease to determine the morbidity and prognosis.Methods A female child was admitted to the hospital on day 20 because of "jaundice for 20 days and difficulty in stopping bleeding at acupuncture sites". Low depth whole genome test of amniocentesis in late pregnancy suggested missing copy number of hemophilia A and X-linked mental retardation type 72. In order to further confirm the diagnosis and prognosis, peripheral blood of the children and their parents were collected for gene testing, chromosome inactivation test and genetic analysis.Results Chromosome Xq28 of the child had 439.4 kb copy number heterozygous deletion variation, which was a clear disease-coding gene for functional loss included in ClinGen database. Chromosome inactivation test showed that the paternal X chromosome of the child was extremely inactivated. Haplotype analysis suggested that the normal chromosome of the subject was inherited from the mother, and there was heterozygous deletion on the paternal X chromosome, so it was inferred that the child will not develop disease or just have mild symptoms.Conclusion It is necessary to analyze the X chromosome inactivation test for female patients with the pathogenic variation of X-linked recessive genetic disease to determine the possibility of the disease.
X连锁隐性(X-linked recessive, XLR) 遗传指的是致病基因位于X染色体上, 其传递方式为隐性, 常见的XLR遗传病包括血友病、红绿色盲、假肥大性肌营养不良等。由于女性有两条X染色体, 当隐性致病基因在杂合状态(XAXa)时, 女性表现为正常的致病基因携带者, 半合子男性则会患病。尽管如此, 女性XLR遗传病患者仍屡见不鲜, 大部分可能与突变或染色体失活等相关。其中染色体失活是指带有野生基因的X染色体失活, 而携带突变基因的X染色体活跃表达。本研究针对1例存在染色体Xq28杂合缺失疑似患有XLR遗传病的女性婴儿进行了相应的遗传学检测及分析, 以判断其发病可能性及评估预后。现针对该研究进行回顾性分析, 以期加强对XLR遗传病染色体失活的认识。
患儿女性, G1P1, 胎龄39+2周, 顺产娩出, 出生体重3.25 kg, 因皮肤黄疸20 d、针刺部位难止血入院。母孕晚期彩超怀疑胎儿发育异常(疑似鼻骨缺失)行羊水穿刺低深度全基因组测定, 提示存在甲型血友病和X连锁智力障碍72型相关拷贝数缺失。体格检查:经皮胆红素值(TCB) 19 mg/dL, 皮肤未见出血点, 前囟平软, 双侧瞳孔等大等圆, 对光反应灵敏, 心肺未见异常。腹软, 肝脾肋下可触及, 质地软, 肠鸣音正常。四肢肌力及肌张力正常, 原始反射可引出。辅助检查提示总胆红素321 μ mol/L, 间接胆红素308 μ mol/L, 直接胆红素13 μ mol/L, 总胆汁酸 17 μ mol/L, 谷草转氨酶76 U/L; 血巨细胞病毒IgG与IgM均阳性, 尿巨细胞病毒DNA定量阳性; 肝胆胰脾彩超示肝脏与脾脏轻度增大; 头颅MRI、彩超及泌尿系彩超未见异常; 耳声发射及脑干听觉诱发电位检查未见异常。凝血功能及凝血因子活性检测如下:纤维蛋白原1.95 g/L, 抗凝血酶71.70%。凝血因子Ⅱ 活性55%(79~131); 凝血因子Ⅷ 活性42.40% (参考值为50%~150%), 凝血因子Ⅸ 活性38.10% (参考值为65%~150%), 凝血因子Ⅹ 活性60.70%(参考值为77%~131%), 凝血因子Ⅺ 活性51.6%(参考值为65%~150%), 凝血因子Ⅻ 活性 48.7%(参考值为50%~150%), 凝血酶原时间 11.70 s(参考值为9.9~12.8 s), 活化部分凝血活酶时间(APTT)48.20 s(参考值为25.1~36.5 s), 国际标准化比值1.08。患儿存在出血倾向, 结合其凝血因子活性轻度下降、APTT延长、孕期羊水基因组测序不能排除血友病A等XLR遗传病, 且家长对患儿羊水穿刺结果极度担忧, 故进一步对该患儿进行遗传学分析以明确诊断及判断预后。研究经本院伦理委员会批准[伦理号:2019-(伦审)-041], 并得到患儿父母的知情同意。
经家属同意, 获取患者外周血进行检测。使用血液基因组DNA提取试剂盒(DP348-03, 天根生化科技有限公司)提取患者外周血中的基因组DNA, 应用Hieff NGS OnePot DNA Library Prep Kit for Illumina试剂盒(翌圣生物科技股份有限公司)和Twist Custom Panel定制探针进行基因文库构建和外显子组捕获。DNBSEQ-T7平台(华大智造)Pair-End 100 bp模式对捕获后文库进行测序。使用BWA软件将测序序列与人类基因组参考序列(GRCh37/hg19版本)进行比对, 对序列进行去重和碱基校准后, 使用GATK软件分析SNP、Indel变异。通过The Human Gene Mutation Database(HGMD)等专业数据库、SIFT等变异有害性生信预测软件以及其他数据库对变异进行注释。此外, 还采用xhmm和clamms工具对探针覆盖区域进行拷贝数变异(CNV)分析。
通过检测雄激素受体基因(AR)的甲基化状态分析X染色体失活(XCI)模式。从患者及其父母的外周血中分离基因组DNA, 应用甲基化敏感限制性内切酶HpaⅡ 消化基因组DNA, 每个样品设置无酶消化对照。将酶切后DNA作为模板扩增MIC2基因上3个HpaⅡ 酶切位点以报告酶切效果。用FAM荧光标记的引物扩增酶切完全DNA和对照DNA, PCR产物包含AR基因上STR位点和2个HpaⅡ 酶切位点。采用毛细管电泳(ABI 3730xl DNA 分析仪)对PCR产物进行检测, 利用GeneMarker V3.0.1软件分析产物长度和峰面积并计算XCI比值。
通过多态性位点分析先证者中携带杂合CNV缺失变异的异常染色体来源。以基因组DNA为模板进行长片段PCR扩增, 以长片段PCR产物为模板, 通过Hieff NGS Fast-Pace DNA Fragmentation Reagent和Hieff NGS Fast-Pace DNA Ligation Module试剂盒(翌圣生物科技股份有限公司)进行文库构建, 并在NextSeq 500测序仪(Illumina公司)对文库进行Single-End 150模式高通量测序。PCR引物位于CNV缺失变异的内部, 产物为正常X染色体上的相应区段, 经gnomAD数据库比对, 该区域内包含多个高频变异位点, 通过对家系成员中多态性位点的基因型分析, 判断先证者缺失变异的遗传来源。
全外显子测序证实受检者Xq28存在约439.4 kb的拷贝数杂合缺失变异, 且为新发变异(父母未检出该变异), 覆盖RAB39B、CLIC2基因全部区域及F8基因编码区约至22号外显子区域(图1), 是ClinGen数据库收录的明确功能缺失致病编码基因(单倍剂量不足评分为3分), 根据ACMG拷贝数变异解读和报告相关指南, 缺失区域包含单倍剂量不足的基因可评为致病基因。RAB39B基因异常可导致精神发育迟滞72型、Waisman综合征, CLIC2 基因异常可导致精神发育迟滞综合征32型, F8基因异常可导致血友病A型, 均为 XLR遗传病。
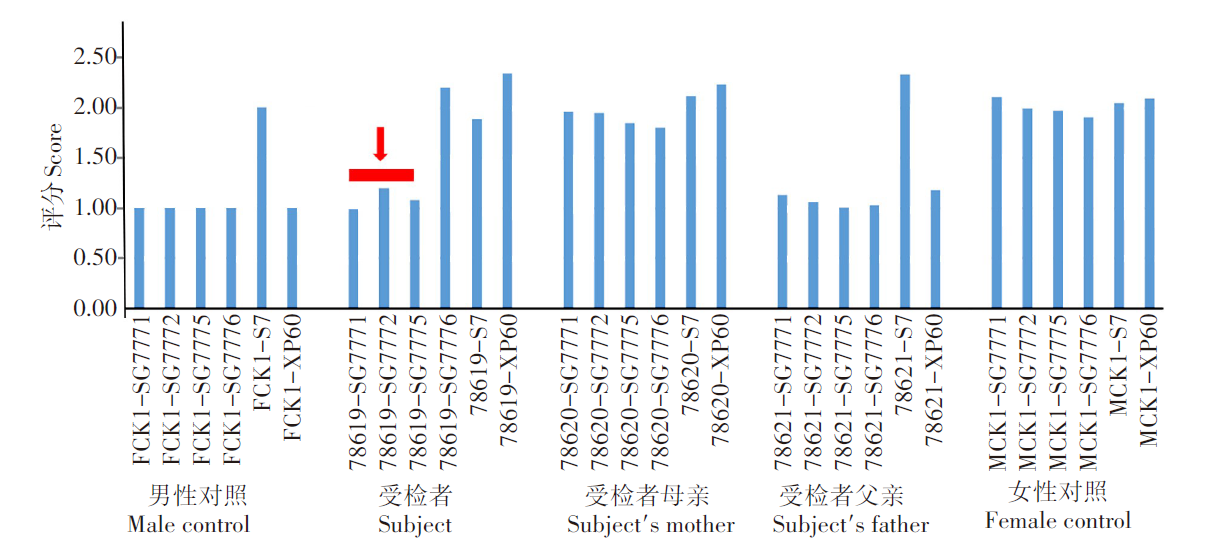 | 图1 先证者家系全外显子测序 受检者 Xq28 存在约 439.4 Kb 的拷贝数杂合缺失变异, 该区域涉及F8、RAB39B、CLIC2 共 3 个已知遗传病致病基因, 且为新发变异。Fig. 1 Whole exon sequencing of the patient’s family Xq28 was found to have a 439.4 Kb copy number heterozygous deletion variant, which involved three genetic disease-causing genes, F8, RAB39B and CLIC2, and was a novel variant. |
染色体失活检测显示患儿父源 X 染色体的失活比例为94.4%, 为极度失活偏移(图2)。单倍型分析结果提示受检者正常染色体遗传自母亲, 父源X染色体上存在杂合缺失(表1)。结合X染色体失活偏倚检测提示父源X染色体失活比例高, 推测该患儿不会表现, 或仅会表现出较轻的疾病症状。
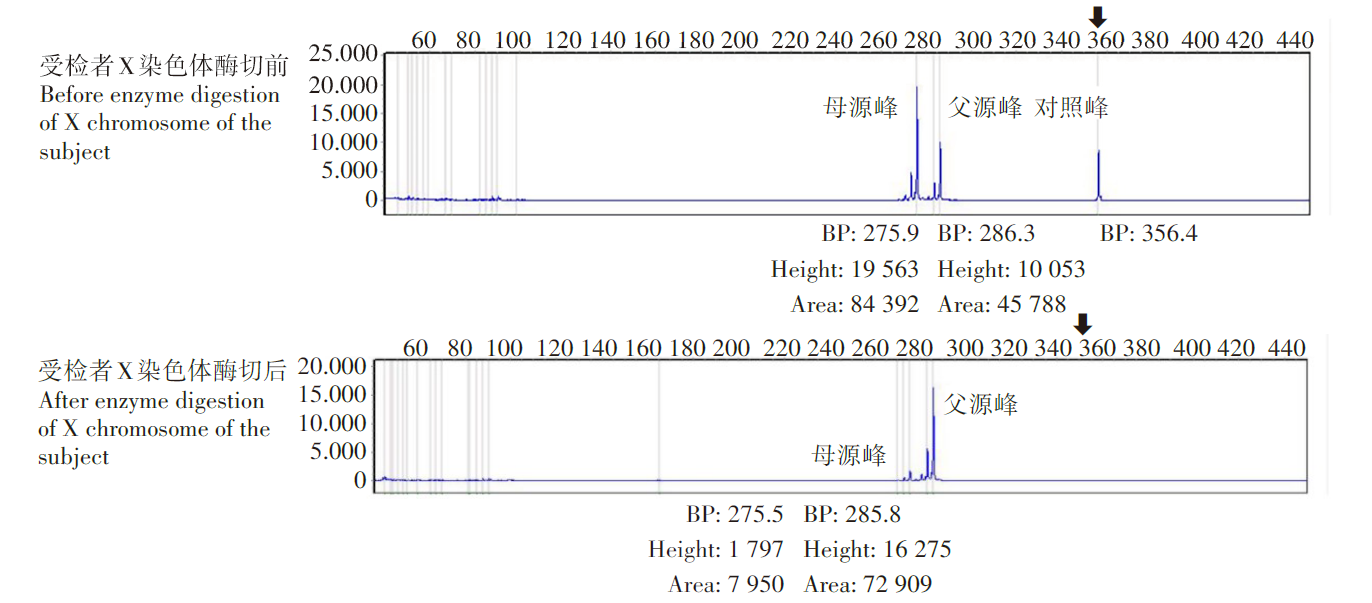 | 图2 染色体失活检测 BP. 扩增产物片段长度; Height. 扩增产物峰高; Area. 扩增产物峰面积; 黑色箭头指示对照峰被完全酶切消化, 提示酶切反应完全; 母源峰大部分被酶切消化, 提示母源X染色体失活比例低; 父源峰基本未被酶切消化, 提示父源X染色体失活比例高。Fig. 2 Detection of chromosome inactivation BP. Amplified product fragment length; Height. The peak of amplification product is high; Area. Enlarge the peak area of yield increase; The black arrow indicates that the control peak was digested completely by enzyme digestion, indicating that the enzyme digestion reaction was complete. Most of the maternal peak was digested by enzyme digestion, suggesting that the proportion of maternal X chromosome inactivation was low. The paternal peak was largely undigested by enzyme digestion, suggesting a high proportion of paternal X chromosome inactivation. |
| 表1 单倍型分析结果 Table 1 Results of haplotype analysis |
治疗及随访情况:患儿入院后给予维生素K1肌注等对症处理及原发病治疗后未再出现针刺部位出血现象, 复查出凝血功能正常。6月龄与12月龄复诊, 患儿均未出现外伤后出血不止或自发出血情况, 血常规及出凝血功能、凝血因子均未见异常。随访神经行为发育正常。证实受检者预后良好, 与研究结论相符合。
本案例为女性患者, 母孕晚期羊水穿刺低深度全基因组测定提示甲型血友病和X连锁智力障碍72型相关拷贝数缺失。由于出生后患儿出现疑似出血倾向及家属的强烈意愿, 故采集患儿及其父母外周血行基因检测。结果提示患儿染色体Xq28 存在约 439.4 Kb 的拷贝数杂合缺失变异, 是ClinGen数据库收录的明确功能缺失致病编码基因。进一步行遗传学分析证实患儿父源 X 染色体为极度失活偏移并存在杂合缺失, 故推断患儿不发病或症状轻微。该研究结论通过对患者的随访得到了进一步证实。
本案例患者外周血全外显子测序证实其Xq28变异区域所累及的血友病A、精神发育迟滞综合征32型、72型等疾病均为XLR遗传病。XLR遗传病是指疾病的致病基因位于X染色体上, 其传递方式为隐性, 患者通常为半合子男性。如出现XLR女性患者则常见以下两种情况:母亲为携带者, 父亲为患者导致女性患者为隐形基因纯合子; 或者, 女性携带者带有正常基因的X染色体出现了X染色体倾斜性失活或X染色体失活偏移(skewed X-chromosome inactivation, SXCI)。本例患儿出现疑似出血倾向、APTT延长及凝血因子下降等, 不能排除是否为女性血友病A患者。女性血友病A极为罕见, 主要表现为关节、肌肉、内脏和深部组织自发性或轻微外伤后出血难止, 常在儿童期起病[1], 偶见文献报道由于SXCI导致女性血友病A的案例[2, 3]以及女性早产儿同时存在Turner综合征及血友病A 的病例报道[4]。由于肝脏是合成凝血因子的主要器官, 如果在肝脏组织中携带正常凝血因子编码基因的X染色体不巧被失活了, 也可能会出现杂合子基因型的女性重型血友病患者[5]。针对该患儿, 新生儿期可能存在生理性凝血因子缺乏, 而且该患儿CMV-IgM阳性, 提示存在巨细胞病毒感染, 也可能导致维生素K合成缺乏引起继发凝血因子下降, 故并不能从临床症状及辅助检查确诊其出血倾向是否为血友病A导致。另外, 精神发育迟滞相关疾病在新生儿期也难以确诊。但是不论血友病或精神发育迟滞, 均需从生命早期开始综合管理, 避免可能出现的并发症并及早进行神经行为发育评估及康复治疗。故针对本例患儿存在的拷贝数杂合缺失变异进行进一步的遗传学分析以判断其致病性有着重要的临床意义。
由于先证者父母既往体健, 全外显子测定均未见异常, 故需进一步评估该先证者是否存在X染色体倾斜性失活。 X染色体失活(X chromosome inactivation, XCI) 是目前已知的表观遗传现象之一。X染色体相比于Y染色体会携带更多的遗传信息, 导致雄性与雌性之间X连锁基因拷贝数差异过大, 因此哺乳动物中一般通过雌性个体(XX)失活一条X染色体实现对雄性(XY)X连锁基因的剂量补偿[6]。女性2条X染色体的其中1条在不同细胞内处于随机失活的状态, 每条X染色体的表达率各为50%, 但是某些情况下 (X失活家族倾向性, 或者细胞选择性生长优势) , 部分女性2条X染色体处于偏性失活状态, 即其中1条X染色体的表达率超过50%甚至接近100%, 称为X染色体倾斜性失活或X染色体失活偏移 [7, 8]。来自父源或者母源X染色体的失活比例> 90%被认为极度失活偏移[9, 10]。失活X染色体上常有不同形式的甲基化, 使其转录受到抑制。由于甲基化敏感性限制性内切酶对含有甲基化碱基的区域不能进行切割, 因此可以基于这一差异特点分析X染色体失活情况。本案例通过检测雄激素受体基因(AR)的甲基化状态分析该患者的XCI模式, 确定父源X染色体的失活比例为 94.4%, 为极度失活偏移。
在XLR遗传病中, 如果女性患者带有野生基因的X染色体失活, 而携带突变基因的X染色体活跃表达, 患者就会表现出相应的致病症状[11]。除了血友病A会出现SXCI导致的女性患者, 其他XLR遗传病也存在类似现象。例如遗传性原发性免疫缺陷疾病慢性肉芽肿病(chronic granulomatous disease, CGD) X染色体连锁隐形遗传占70%, 通常为男性患者, 女性携带者表现为健康状态, 但是SXCI现象会导致极少部分青少年及成年女性携带者出现临床症状, 甚至婴儿期起病[12]。同样为XLR遗传病的Wiskott-Aldrich综合征也存在由于SXCI导致的女性患者[13]。本例先证者存在SXCI, 故需进一步分析先证者中携带杂合CNV缺失变异的异常染色体来源。该研究通过多态性位点分析, 证实缺失片段正好位于失活X染色体上, 因此推测该患儿不会出现相应疾病表型或仅症状轻微。随访结果也进一步证实了该推测的正确性。
目前X染色体失活的机理仍不明确。已有研究发现X染色体失活中心(X chromosome inactivation center, XIC)是XCI主要的调控区域。X-非活性特异性转录物(X-inactive specific transcript, XIST)位于XIC中, 是调控XCI的主要基因, 其表达产物为XIST RNA, 长度为15~17 kb, 是一种顺式作用的RNA, 不会进一步翻译为蛋白质。XIST 作为一个长非编码RNA是X染色体失活的分子基础, XIST RNA沿着失活的X染色体呈点状分布, 被覆盖的X染色体区域失去活性[14], 而且XIST RNA会吸引和富集已知的大约100种蛋白到X染色体上, 并进一步稳定和维持X染色体的失活[5]。另外, 染色体失活可能与DNA甲基化也有关联。DNA甲基化是重要的表观遗传基因修饰, 参与了许多表观遗传活动, 在失活的X染色体上, 很多区域的DNA被甲基化。但是2条X染色体都具有XIST基因, 为什么只有其中1条去甲基化导致其所在的X染色体失活, 以及为什么XIST基因的转录产物表现为顺式作用, 机理仍不清楚。另外, 研究证实XCI在哺乳动物中具有物种特异性, 但目前仍缺乏理想的系统来研究人早期胚胎发育过程中的X染色体调控机制[15]。虽然大部分情况下, 染色体的偏倚失活为随机失活, 但是也有研究发现如果患者的染色体存在涉及到X染色体的缺失、重复或等臂染色体等非平衡结构畸变, 通常结构异常的X染色体优先失活, 因此部分研究者推测生命本身倾向选择结构畸变的 X 染色体失活以降低该畸变可能导致的不良后果[14]。关于这一点也有不同意见, 有的研究提示失活X染色体在细胞核内经历了重大的构象重组, 因此才导致其功能和结构的不同[16]。X染色体失活的机理仍有待于进一步研究。
综上所述, X染色体失活的研究对于XLR遗传病患者进行早期诊断或排除都具有重要的意义。由于X染色体失活检测是针对DNA进行的试验, 可以考虑通过从羊水或脐带血对疑似XLR遗传病的胎儿细胞DNA进行X染色体失活的遗传学分析。对于遗传学检测发现存在XLR遗传疾病致病性变异的女性患者有必要进一步明确其是否存在SXCI以及SXCI与致病性变异的关系, 从而判断其发病的可能性, 达到早期防治及遗传咨询的目的。
利益冲突声明 所有作者声明不存在利益冲突
编辑:王佳燕
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|