
{kind=link}
11个儿童线粒体脑肌病家系的临床分析
[胡璐, 逯军*  ]
]
]
|
|


作者简介:胡璐(1996—),女,硕士研究生,研究方向:儿童线粒体疾病。
目的 探讨儿童线粒体脑肌病(mitochondrial encephalomyopathy, ME)家系的临床表型及基因型特征。方法 回顾分析2007年1月至2021年12月就诊于海南省3家三甲医院儿科的11个ME家系的临床资料及基因检测结果。结果 11个ME家系中共确诊13例患儿,其中男性患儿6例(46.15%),女性患儿7例(53.85%)。发病年龄为6个月~12岁,从发病至确诊病程间隔9个月~8年,Morava评分为6~11分。临床症状主要有运动异常、发育落后或倒退、癫痫发作、卒中样发作;13例患儿中血乳酸升高11例(84.62%),血肌酸激酶升高4例(30.77%);头颅MRI主要累及颞顶枕叶、小脑、脑干及基底节区,部分伴有脑萎缩;基因检测出8个家系为mtDNA突变致病(72.72%),其中5个家系、6例患儿为 MT-TL1基因m.3243 A>G,且有4个家系(80.00%)检测出该位点的无症状突变携带者5例;2个家系、3例患儿为 MT-ND5基因m.13513 G>A,且有1个家系(50.00%)检测出该位点的无症状突变携带者1例;1个家系、1例患儿为 MT-ND3基因m.10191 T>C位点,且检测出该位点的无症状突变携带者2例。3个家系为nDNA突变致病(27.27%),其中1个家系、1例患儿为 SURF1基因c.751C>T,c.516-2A>G位点的复合杂合变异,遵循常染色体隐性遗传,致病位点分别遗传自母亲及父亲;2个家系、2例患儿分别为 DNM1L基因的两个自发新发突变c.1040C>G、c.2060_2062delTAG。所有患儿经基因检测确诊后均给予线粒体鸡尾酒疗法及对症治疗,随访至2022年6月,其中有2个家系失访,定期随访的9个家系、11例患儿中,尚有3例存活。结论 对确诊ME的患儿,完善家系成员的基因检测可筛查出早期无症状的致病突变携带者,实现ME的早诊断并指导临床遗传咨询;研究发现了 DNM1L基因的2个新发致病位点,扩展了基因型谱。
Objective To investigate the clinical phenotype and genotype characteristics of mitochondrial encephalomyopathy (ME) families in children.Methods The clinical data and genetic test results of eleven ME families who were admitted to the department of pediatrics of three tertiary hospitals in Hainan Province from January 2007 to December 2021 were retrospectively analyzed.Results A total of 13 cases were diagnosed in eleven ME families, including 6 males (46.15%) and 7 females (53.85%). The age of onset ranged from 6 months to 12 years, the interval from onset to diagnosis was 9 months to 8 years and Morava score was 6-11. Clinical symptoms mainly included abnormal movement, developmental retardation or regression, seizures, stroke-like episodes; among the 13 children, 11 (84.62%) had elevated blood lactic acid and 4 (30.77%) had elevated blood creatine kinase. Cranial MRI mainly involved temporal parietal occipital lobe, cerebellum, brainstem and basal ganglia, some with brain atrophy. Gene detection showed that 8 families (72.72%) were caused by mtDNA mutation, of which 5 families and 6 patients were caused by MT-TL1, m.3243A>G, and 5 asymptomatic carriers of 4 families (80.00%) were detected; MT-ND5, m.13513 G>A was detected in 2 families and 3 patients, and an asymptomatic mutation carrier was detected in a family (50.00%); MT-ND3, m.10191T>C was detected in one family and one patient, and 2 asymptomatic mutation carriers were detected. Three families were caused by nDNA mutations (27.27%). A compound heterozygous mutation of c.751C>T and c.516-2A >G in SURF1 gene was found in one family and one patient, which followed autosomal recessive inheritance. The pathogenic loci were inherited from mother and father, respectively. Two new spontaneous mutations c.1040C>G and c.2060_2062delTAG in DNM1L gene were respectively detected in two families and two patients. All children were given mitochondrial cocktail therapy and symptomatic treatment after diagnosis by genetic testing. Follow-up to June 2022, two families were lost to follow-up and 9 families were followed up regularly; three of the 11 children were still survived.Conclusions For children diagnosed with ME, genetic testing of family members can screen out early asymptomatic pathogenic mutation carriers, achieve early diagnosis of ME and guide clinical genetic counseling. Two new pathogenic sites of DNM1L gene were found in this study, which expanded the genotype spectrum.
线粒体脑肌病(mitochondrial encephalomyopathy, ME), 泛指一组由线粒体基因(mitochondrial DNA, mtDNA)或细胞核基因(nuclear DNA, nDNA)发生突变导致线粒体结构和功能异常, 以脑和肌肉受累为主要临床表现的多系统受累的罕见遗传性疾病, 2018年5月11日, 我国卫生健康委员会等5部门联合制定了《第一批罕见病目录》, ME被收录其中[1]。2016年GORMAN等[2]的研究提示在小于16岁的儿童中ME患病率为5/10万~15/10万。英国北部一项筛查研究也表明, 新生儿中10种常见的致病性mtDNA点突变携带率达1/200, 其中m.3243 A> G点突变率最高[3], 目前我国ME患病率尚不明确。自1981年ANDERSON[4]测定了人类mtDNA的完整基因组序列至今, 伴随着基因分子遗传学诊断手段的发展, 人类对该ME遗传学病因的研究逐步深入; 但由于ME临床表型的复杂多样性和异质性, 目前临床诊治过程中仍存在相当高比例的漏诊、误诊及误治[5]。
家系分析是了解、研究及诊断遗传罕见病的重要手段, 从先证者入手, 根据对其家族成员的发病情况调查绘制家系图, 根据遗传罕见病的家族聚集性及其传递规律判断遗传方式, 对疾病的致病性分析、遗传咨询等均具有积极的临床意义。本研究以家系为研究单位, 通过总结11个ME先证者患儿家系的临床表型、基因型特征及诊治过程, 旨在为临床医师提供ME早期识别及诊治经验。
对2007年1月至2021年12月就诊于海南省3家三甲医院儿科的11个ME先证者患儿家系的临床资料及基因检测结果进行分析。本研究已通过中南大学湘雅医学院附属海口医院伦理委员会审核和批准, 伦理审查批件编号:2021-(伦审)-040, 患儿法定监护人签署知情同意书。
1.1.1 纳入标准 符合以下所有条件:①年龄0~14岁; ②基因检测提示确诊ME; ③基因检测提示可能致病、结合临床表型及致病性分析可确诊为ME。
1.1.2 排除标准 符合以下任一条件:①年龄> 14岁; ②基因检测排除ME; ③未行基因检测; ④未行家系验证; ⑤未取得患儿法定监护人知情同意书。
1.2.1 临床资料收集 收集患者一般信息、现病史、家族史、体格检查、辅助检查、Morava评分、基因检测及治疗、随访、预后情况等。
1.2.2 基因检测结果收集 包括基因检测方法、基因突变方式、突变位点及致病性评级结果, 若为mtDNA突变, 收集突变异质性。
使用SPSS26.0及Microsoft Office Excel 2019软件进行数据处理。
本研究共收集11个ME先证者患儿家系, 共确诊13例患儿, 其中男性患儿6例(46.15%), 女性患儿7例(53.85%), 男女比例约为0.86∶ 1; 发病年龄最小6个月, 最大12岁; 从发病至确诊病程间隔约9个月~8年; Morava评分处于6~11分之间, 平均为(8.23± 1.36)分。见表1。
| 表1 11个家系线粒体脑肌病患儿的临床资料 Table 1 Clinical data of 11 families with mitochondrial encephalomyopathy |
首发症状:癫痫发作6例(46.15%), 运动异常5例(38.46%), 发育落后或倒退3例(23.08%), 头痛2例(15.38%), 胃肠道症状2例(15.38%), 眼部症状2例(15.38%), 记忆力减退1例(7.69%)。确诊时主要临床症状:运动异常8例(61.54%), 发育落后或倒退8例(61.54%%), 癫痫发作7例(53.85%), 卒中样发作3例(23.08%), 眼部症状3例(23.08%), 听力下降2例(15.38%), 胃肠道症状2例(15.38%), 内分泌受累1例(7.69%)。
2.3.1 血乳酸及肌酸激酶结果 所有患儿均行血乳酸及肌酸激酶测定, 11例(84.62%)血乳酸升高, 平均值(4.55± 2.75) mmol/L(参考值范围:小于1.7 mmol/L); 4例(30.77%)血肌酸激酶升高, 平均值(193.85± 204.60) IU/L(参考值范围:< 190 IU/L)。
2.3.2 头颅MRI结果 所有患儿均行头颅MRI检查, 累及顶叶6例(46.15%), 累及枕叶6例(46.15%), 累及颞叶4例(30.78%), 伴有脑萎缩3例(23.08%), 累及小脑3例(15.38%), 累及脑干2例(15.38%), 累及基底节区2例(15.38%), 累及放射冠2例(15.38%), 累及大脑脚2例(15.38%), 累及丘脑1例(7.69%)。
5个家系(45.45%)行线粒体全基因组测序, 3个(27.27%)家系行线粒体全基因组测序及全外显子组测序(WES), 2个(18.18%)家系行聚合酶链反应-限制性片段长度多态性分析技术检测(PCR-RFLP), 1个(9.09%)家系行线粒体全基因组测序及多重连接探针扩增检测(MLPA); 基因检测出8个家系的致病变异为mtDNA突变(72.72%), 其中5个家系、6例患儿为MT-TL1基因m.3243 A> G位点, 且有4个家系(80%)检测出该位点的无症状突变携带者6例; 2个家系、3例患儿为MT-ND5基因m.13513 G> A位点, 且有1个家系(50%)检测出该位点的无症状突变携带者1例; 1个家系、1例患儿为MT-ND3基因m.10191 T> C位点, 且检测出该位点的无症状突变携带者2例。3个家系的致病变异为nDNA突变(27.27%), 其中1个家系、1例患儿为SURF1基因c.751C> T, c.516-2A> G位点的复合杂合变异, 遵循常染色体隐性遗传, 致病位点分别遗传自母亲及父亲; 2个家系、2例患儿分别为DNM1L基因的两个自发新发突变:c.1040C> G、c.2060_2062delTAG, 经Sanger测序验证父母均未发现相应变异(PS2 ), 上述两个变异在ESP数据库、千人数据库、EXAC数据库的正常对照人群中未发现(PM2), c.1040C> G突变位点经多个生物信息软件预测可对基因或基因产物造成有害影响(PP3), c.2060_2062delTAG突变位点为非重复序列区域框内缺失变异、可引起的蛋白质长度改变(PM4), 根据美国医学遗传学与基因组学学会(The American College of Medical Genetics and Genomics, ACMG)指南, 本研究中两个新发突变位点致病性等级均为可能致病性变异(PS2+PM2+PP3, PS2+PM2+PM4); 并且这2例患儿临床表型与既往报道病例高度符合, 综合临床表型及基因型判定上述两个自发新发突变为患儿的致病原因, 是DNM1L基因的两个新发致病位点。见表2、图1。
| 表2 11个家系线粒体脑肌病患儿的基因检测结果 Table 2 Genetic detecting of 11 families with mitochondrial encephalomyopathy |
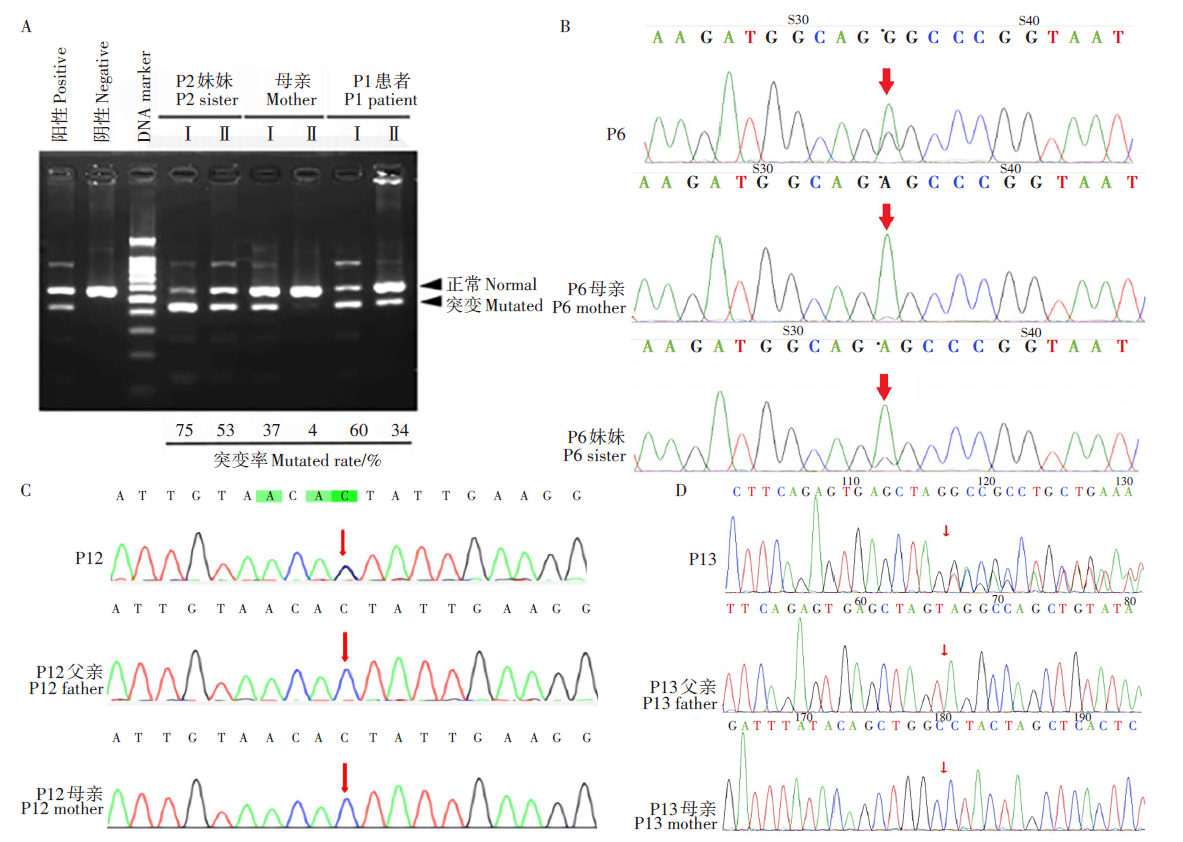 | 图1 部分家系基因检测图 A. 家系1聚合酶链反应一限制性片段长度多态性分析技术检测结果图, Ⅰ .尿液沉淀细胞中的mtDNA突变异质性, Ⅱ .血细胞中的mtDNA突变异质性; B. 家系5线粒体全基因组测序结果图; C. 家系10全外显子组测序结果图; D. 家系11全外显子组测序结果图; B~D中箭头表示相应家系的基因突变位点。Fig. 1 Gene detection pictures of some families A. Picture of PCR-RFLP result in family 1 , Ⅰ . heterogeneity of mtDNA mutation in urine sediment cells, Ⅱ . heterogeneity of mtDNA mutation in blood cells; B. Picture of whole mitochondrial genome sequencing result in family 5; C. Picture of whole exome sequencing result in family 10; D. Picture of whole exome sequencing result in family 11; Arrows in picture B to D indicate the the gene mutation sites of the relevant family. |
11个家系的先证者患儿经基因检测确诊后均给予线粒体鸡尾酒疗法, 用药方案为复合辅酶或辅酶Q10或三磷酸腺苷二钠联合左卡尼汀、维生素B1片、维生素B6片、维生素C片、维生素E片、甲钴胺片、L-精氨酸、叶酸及肌苷片等治疗, 其中患儿P2、P8在家系基因筛查后提前给予线粒体鸡尾酒疗法干预治疗; 患儿P9曾接受异体干细胞移植治疗; 其余给予抗癫痫、护胃、营养心肌、控制血糖及康复训练等对症治疗。随访至2022年6月, 有2个家系(患儿P11、P13)已失访, 其余患儿定期门诊或电话随访, 因呼吸道、泌尿道等感染或因出现新的系统受累导致病情加重后于我科多次住院治疗; 截至2022年6月, 定期随访的9个家系、11例患儿中, 尚有3例(P4、P5、P12)存活, 其余8例因重症感染、呼吸衰竭、消化道大出血、慢性肾功能不全、全身多器官功能衰竭等病因已死亡, 其中患儿P9接受移植后出现移植物抗宿主反应, 导致病情加重, 最终死于多器官功能衰竭。本研究中患儿生存时间为10个月~11年, 平均约3年5个月。
线粒体是真核细胞内通过氧化磷酸化过程产生大量三磷酸腺苷(ATP)的具有半自主复制能力的细胞器, 被称为细胞的氧化中心、动力工厂, 其双链环状mtDNA分子编码37个基因, 和nDNA共同参与线粒体的各项生命活动。线粒体脑肌病是一组由于mtDNA或nDNA突变、导致线粒体结构或功能异常的遗传罕见病, 主要累及脑、肌肉等能量需求较高的组织。ME的具体发病机制尚不十分清楚, 现有研究认为可能与基因突变所致的线粒体氧化呼吸链复合体亚基缺陷、mtDNA翻译缺陷、线粒体蛋白质组装缺陷、线粒体内膜结构异常、以及线粒体动力学障碍等有关[6, 7, 8, 9]。该病可发生在各个年龄, 可累及全身多组织、器官或系统, 常见的临床表现主要有抽搐或癫痫发作、精神发育迟滞、认知障碍、偏头痛、卒中样表现、共济失调等中枢神经系统损害, 肌无力、肌阵挛、运动不耐受等骨骼肌损害, 视野缺损、眼球活动障碍、听力下降甚至耳聋、身材矮小、心肌病、胃肠道症状、内分泌代谢异常、周围神经病等全身多系统损害, 临床表型复杂多样、异质性高, 病程可呈急性或缓慢进展。目前常见的ME主要包括线粒体脑肌病、高乳酸血症和卒中样发作综合征(mitochondrial encephalomyopathy, lactate acidosis and stroke-like episodes, MELAS)、Leigh综合征(Leigh's syndrome, LS)、肌阵挛性癫痫伴破碎红纤维病、 Leber遗传性视神经病等。本研究共收集了5个MELAS家系, 4个LS家系及2个线粒体和过氧化物酶体裂变缺陷致脑病1型家系(encephalopathy due to defective mitochondrial and peroxisomal fission-1, EMPF1)。
ME的经典诊断思路是基于详细的病史及体格检查总结出相关的临床表型, 其次完善血和(或)脑脊液乳酸、肌酸激酶、丙酮酸、乳酸/丙酮酸等生化检测、肌肉活检后行呼吸链酶复合物活性测定及组织免疫化学染色等实验室检查, 头颅MRI及波谱分析等影像学检查; 同时作为一组罕见遗传病, 基因检测是诊断ME的“ 金标准” [10, 11]。根据基因分子诊断结果, ME可分为两类:一类是由nDNA突变致病, 遵循孟德尔遗传模式, 另一类是由mtDNA突变致病, 遵循母系遗传模式。文献指出80%~90%儿童线粒体疾病是由nDNA突变致病的[12], 本研究共检测出8个mtDNA突变致病的家系, 占比72.72%, 3个nDNA突变致病的家系, 占比27.27%, 这主要与ME患病率低、本研究纳入家系数相对较少有关。同时由于mtDNA突变致病的ME存在母系遗传、异质性、阈值效应及瓶颈效应等疾病特征, 其临床表型和基因型的关联通常不是直接对应, 即不同基因的变异可能导致相同的临床表型, 同一基因的变异亦可能导致不同的临床表型[13, 14], 异质性水平的高低是导致不同患儿间或同一家系不同个体间临床表型差异的主要原因之一, 本研究中8个mtDNA突变致病的家系共检测出ME患儿10例, 其突变异质性波动为34%~75%, 无症状突变携带者8例, 其突变异质性波动为4%~30%, 这主要与阈值效应, 即mtDNA突变异质性水平必须超过组织特异性阈值水平才会出现临床表型有关。
同时为了便于诊断, 2002年, BERNIER等[15]初步建立了线粒体呼吸链缺陷相关疾病的评分标准, 2006年MORAVA等[16]将该评分标准进一步简化为临床疑诊线粒体疾病儿童的初步筛查工具, 即Morava评分系统。该评分系统主要根据上述ME经典诊断思路中肌肉、中枢神经系统和其他多系统的临床症状、代谢和影像学结果以及肌肉病理活检结果进行评分:1分提示不太可能为线粒体疾病, 2~4分提示可能为线粒体疾病, 5~7分提示很可能为线粒体疾病, ≥ 8分提示明确为线粒体疾病。近年来该评分系统仍被用于临床诊治过程中, WITTERS等[17]进行的一项多中心的回顾性研究指出Morava评分体系有助于线粒体疾病的临床诊断, 阳性预测值为69.5%, 阴性预测值为38.9%, 并且指出nDNA突变患者的评分比mtDNA突变患者更高, 这在一定程度上也可协助全外显子测序结果的解读。我国学者对56例临床疑诊为ME患者的临床研究表明使用Morava评分系统进行ME的初筛, 可避免漏诊[18]。本研究中13例患儿的Morava评分均提示很可能或明确为ME, 其中mtDNA突变的患儿P1及P3曾进行了肌肉活检, 为Morava评分提供了更多依据, 使其评分略高于nDNA突变患儿。肌肉活检是一项侵入性检查, 并且儿童患者因依从性差、通常需先进行麻醉, 这对ME患儿存在一定的风险和有害性[19], 并且肌肉活检结果尚不能区分原发性和继发性线粒体功能障碍[20], 因此在本研究应用少。此外研究者们发现了与线粒体功能障碍相关的新型生物标志物:成纤维细胞生长因子21及分化生长因子15, 具体诊断价值仍需进一步临床实践验证[21, 22]。
目前ME仍无标准有效的治愈方法, 临床常用治疗方案依旧为经典的线粒体鸡尾酒疗法, 即复合辅酶或辅酶Q10或三磷酸腺苷二钠联合左卡尼汀、维生素B1片、维生素B6片、维生素C片、维生素E片、甲钴胺片、L-精氨酸、叶酸及肌苷片等治疗, 同时针对其余受累系统进行相应的对症支持治疗, 比如有氧耐力康复训练被证明可以增加线粒体质量、线粒体中酶活性和肌肉的力量[23]。近年来随着对ME致病机制的深入研究, 研究者开发了数种新药:比如人工合成的三萜化合物Omaveloxolone, 该化合物可阻止机体内具有抗氧化作用的核转录因子E2相关因子-2(Nrf2)的降解, 2020年MADSEN等[24]曾在53例ME患者中进行了一项多中心双盲、随机、安慰剂对照试验证明该药具有一定的疗效; 基因治疗呼吸链复合物ND4亚基的异位表达方法已被应用于临床试验[25]。ME通常预后不佳, 患者生存周期短, 因此对已生育有确诊ME患者的家系, 建议进行遗传咨询并接受生殖遗传技术的干预以降低后代患病风险、实现优生优育。目前常用的方法有胚胎植入前基因诊断(preimplantation genetic diagnosis, PGD)或产前诊断, 这两项技术已逐渐被用于预防线粒体疾病。PGD是指从早期胚胎中取出一个或多个细胞(如卵裂期胚胎的卵裂球活检、囊胚滋养层细胞活检和极体活检等)进行基因检测, 选择无突变或低突变的胚胎移植到子宫的技术; 产前诊断是一种在早期妊娠不同阶段对孕母绒毛细胞或羊膜细胞进行的mtDNA突变水平测定的技术, 若mtDNA突变水平高时可选择终止妊娠。此外2016年已有应用基于细胞核移植基础上的线粒体置换技术生育健康儿童的报道, 但涉及伦理及安全性问题, 该项技术的临床应用在国际上尚未普遍获得立法[26]。
综上所述, 临床上对有运动异常、癫痫发作、发育落后或倒退、卒中样发作, 合并眼部、听力、胃肠道等系统受累及高乳酸血症的患儿, 需早期考虑ME并进行Morava评分, 完善家系成员的基因检测可早期筛查出无症状致病突变携带者。近年来随着第二代测序技术的广泛应用, ME的分子诊断水平逐步提高, nDNA及mtDNA的致病变异不断被发现, 为ME新型治疗药物及技术的研发提供了新的思路和方向, 早发现、早诊断、早干预及进行遗传咨询仍是以ME为代表的儿童罕见遗传病临床诊疗的关键步骤。
利益冲突声明 所有作者声明不存在利益冲突
编辑:王佳燕
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|